


ЮЛжУЃКжївГ > МВВЁеяСЦ > ОЋОШОвХ > вХДЋВЁ
ЛљвђЭЛБфжТУИЛюадвьГЃ
РДдДЃК39ПЕИДЭј зїепЃК39ПЕИДЭј ЕуЛї:ДЮ ЪБМфЃК2008-02-24
ЕкШ§НкЁЁЛљвђЭЛБфжТУИЛюадвьГЃ
ЁЁЁЁгЩгкЛљвђЭЛБфЕМжТУИЛюадНЕЕЭЛђдіИпЫљв§Ц№ЕФМВВЁГЦЮЊвХДЋадУИВЁЃЈhereditary enzymopathyЃЉЁЃвХДЋадУИВЁгыЗжзгВЁЕФЧјБ№дкгкКѓепв§Ц№ЛњЬхЙІФмеЯАЪЧЕААзжЪЗжзгБфвьЕФжБНгКѓЙћЃЛЖјЧАепдђгЩгкКЯГЩУИЕААзНсЙЙвьГЃЛђЕїПиЯЕЭГЭЛБфКѓЕМжТУИЕААзКЯГЩЪ§СПМѕЩйЃЌЭЈЙ§УИЕФДпЛЏзїгУМфНгЕМжТДњаЛЮЩТвЫљв§Ц№ЕФЛњЬхЙІФмеЯАЁЃЛљвђЭЛБфЕМжТУИЕФвХДЋБфвьПЩБэЯжЮЊУИЛюадНЕЕЭЁЂУИЛюаде§ГЃЃЈЭЌвхЭЛБфЛђЭЛБфВПЮЛВЛгАЯьУИЛюаджааФЃЉМАУИЛюаддіИпЁЃОјИЮгВЛЏЁЂАзФкеЯЁЂжЧСІЗЂг§ВЛШЋЕШжЂзДЁЃШщРрКЌгаШщЬЧЃЌЫќОЯћЛЏЕРШщЬЧУИЗжНтВњЩњЦЯЬбЬЧМААыШщЬЧЃЌАыШщЬЧЭЈЙ§вЛЯЕСаУИДйЗДгІВњЩњЦЯЬбЬЧЖјБЛзщжЏРћгУЃЈЭМ4-22ЃЉЁЃ
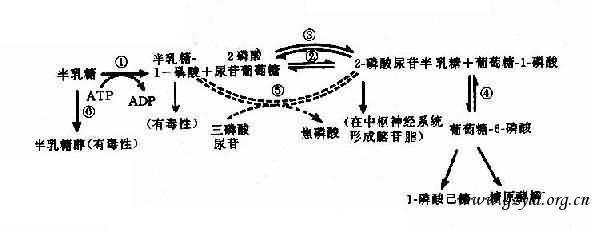
ЭМ4-22 АыШщЬЧДњаЛЭООЖ
ЃЈащЯпБэЪОФъГЄвдКѓВХЗЂеЙЦ№РДЕФДњГЅЭООЖЃЉ
ЂйАыШщЬЧМЄУИЃЛЂкАыШщЬЧ-1-СзЫсФђмеѕЃзЊвЦУИЃЛЂлАыШщЬЧФђме-2-СзЫс-4-вьЙЙУИЃЛЂмСзЫсЦЯЬбЬЧБфЮЛУИЃЛЂн-СзЫсФђмеАыШщЬЧЃЈЛђЦЯЬбЬЧЃЉНЙСзЫсЛЏУИЃЛЂоШЉЬЧЛЙдУИ
ЁЁЁЁЕфаЭЕФАыШщЬЧбЊжЂЛМепгЩгкАыШщЬЧ-1-СзЫсФђмеѕЃзЊвЦУИЃЈgalactose-1-phosphate uridy1 transferase,МђГЦзЊвЦУИЃЉШБЗІЃЈЭМ4-22ЂкЃЉЃЌжТЪЙАыШщЬЧ-1-СзЫсЃЈGal-1-PЃЉМААыШщЬЧЛ§ОлдкбЊжаЃЌВПЗжЫцФђХХГіЁЃGal-1-PдкИЮЕФЛ§ОлПЩв§Ц№ИЮЙІФмЫ№КІЃЌЩѕжСИЮгВЛЏЃЛдкФдЕФЛ§Олв§Ц№жЧСІеЯАЃЛбЊжаАыШщЬЧЩ§ИпПЩЪЙЦЯЬбЬЧЪЭГіМѕЩйЃЌГіЯжЕЭбЊЬЧжЂЁЃАыШщЬЧдкШЉЬЧЛЙдУИзїгУЯТВњЩњАыШщЬЧЃЌФмИФБфОЇзДЕФЩјЭИбЙЃЌЪЙЫЎЗжНјШыЃЌгАЯьОЇзДЬхДњаЛЖјжТАзФкеЯЁЃ
ЁЁЁЁЛМепЖМЪЧвўадДПКЯзгЃЈggЃЉЃЌдгКЯзгБэаЭе§ГЃЃЌзЊвЦУИЛюаддМЮЊ50%ЃЌЛюадЕЭгк10%ПЩГіЯжЕфаЭжЂзДЁЃ
ЁЁЁЁСэвЛРрАыШщЬЧбЊжЂЮЊАыШщЬЧМЄУИЃЈgalactokinaseЃЉШБЗІЫљв§Ц№ЃЌжЂзДНЯЧсЃЌжївЊБэЯжЮЊЧрФъаЭАзФкеЯЃЌбЊжаАыШщЬЧдіИпЃЌЕЋЮоИЮМАФдЫ№КІЁЃ
ЁЁЁЁзЊвЦУИЛљвђЃЈGALLЃЉЖЈЮЛгк9p13ЃЌАыШщЬЧМЄУИЛљвђЃЈGALKЃЉЖЈЮЛгк17q21-q22ЁЃСНВЁОљЮЊГЃШОЩЋЬхвўадвХДЋЁЃ
ЁЁЁЁЃЈ2ЃЉУИШБЗІжТДњаЛЕзЮяЖбЛ§в§Ц№ЕФМВВЁЃКЕБвЛЯЕСаЩњЛЏЗДгІПЩФцЪБЃЌвЛДІЕФзшЖЯГЃЕМжТДњаЛЕзЮяЃЈS1ЃЉжќЛ§ЁЃжќЛ§ЕФЮяжЪШчЙћШмНтЖШИпЃЌдђдкбЊКЭФђжаХЈЖШдіИпЃЛШєШмНтЖШЕЭЪБЃЌдђдкзщжЏжажќЛ§в§Ц№МВВЁЁЃ
ЁЁЁЁЬЧджќЛ§жЂЃЈglycogen storage liseaee,GSDЃЉЪЧвЛзщгЩЬЧдКЯГЩКЭНЕНтУИШБЯнв§Ц№ЕФМВВЁЃЌжСЩйга12жжРраЭЁЃЬЧджќЛ§жЂжївЊРлМАИЮЛђМЁШтЃЌЕЋгаЕФвВПЩАщгааФЁЂЩіКЭЩёОЯЕЭГЕФЫ№КІЁЃВЛЭЌРраЭжЎМфЦфбЯжиадКЭдЄКѓЖМВЛШЋЯрЭЌЁЃР§ШчЃЌvon GirkeВЁЃЈЂёаЭЃЉжЂзДЗЧГЃбЯжиЃЌЖјHersВЁЃЈЂіаЭЃЉНЯЧсЁЃДЫРрМВВЁЗЂВЁЛњРэПЩгУvon GierkeВЁЮЊР§ЫЕУїЁЃБОВЁЪЧгЩгкИЮФкЦЯЬбЬЧ6-СзЫсУИЃЈglucose-6-phosphatase,G6PaseЃЉШБЗІв§Ц№ЁЃИЮЬЧддквЛЯЕСаУИЕФзїгУЯТЩњГЩЦЯЬбЬЧЃЌет ЗДгІЕФИїИіВНжшЖМЪЧПЩФцЕФЃЌЦфжївЊВНжшШчЯТЃК
ЗДгІЕФИїИіВНжшЖМЪЧПЩФцЕФЃЌЦфжївЊВНжшШчЯТЃК
![]()
ЁЁЁЁЛМепгЩгкG6PaseШБЗІЃЌЫљвдG6PВЛФмзЊБфЮЊЦЯЬбЬЧЙЉзщжЏРћгУЃЌЭЈЙ§ПЩФцЗДгІЖјКЯГЩЙ§ЖрЕФИЮЬЧдЃЌв§Ц№ЛМЖљИЮжзДѓЁЃЕБВЛНјЪГЪБМЋвзЗЂЩњЕЭбЊЬЧЁЃгЩгкЖЏгУжЌЗОПЩвдГіЯжЭЊбЊжЂЁЃG6PЭЈЙ§ЮобѕНЭНтЃЌЩњГЩДѓСПШщЫсЃЌЕМжТЫсжаЖОЁЃЫљвдЛМепЕФИЮДѓАщЕЭбЊЬЧЃЌЗЂг§ВЛСМЃЌЯћЪнЃЌЩэЬхАЋаЁЃЌГЃгаГібЊЧуЯђЁЃИЮЛюМьМћЬЧдКЌСПдіМгЁЃ
ЁЁЁЁБОВЁГЪШОЩЋЬхвўадвХДЋЁЃЛМепG6PУИЭъШЋШБЗІЃЌЖрЪ§ЛМепИИФИБэаЭе§ГЃЃЌЕЋG6PУИЛюадЮЊжаМфжЕЁЃЭЌАћжаПЩГіЯжЛМепЁЃ
ЁЁЁЁЃЈ3ЃЉУИШБЗІжТДњаЛжеВњЮяШБЗІв§Ц№ЕФМВВЁЃКетЪЧвЛРргЩE2-3УИШБЗІЃЌжТЫљгаВњЮя(АќРЈжеВњЮяP)ШБЗІв§Ц№ЕФМВВЁ.АзЛЏВЁОЭЪЧЪЕР§.АзЛЏВЁ(albinism)ЗжШЋЩэаЭМАОжВПаЭ.ШЋЩэаЭГЃМћ,ЛМепЦЄЗєГЪАзЩЋ,УЋЗЂвјАзЛђЕЛЦЩЋ,КчФЄМАЭЋПзГЪЕКьЩЋ,ЪгЭјФЄЮоЩЋЫи,апУї,блЧђе№ВќЕШ,БОВЁЗЂВЁТЪдМ1/10000-1/20 000,ГЪГЃШОЩЋЬхвўадвХДЋ.
ЁЁЁЁе§ГЃШЫКкЩЋЫигЩКкЫиЯИАћКЯГЩ,етаЉЯИАћжагаЬиЪтЕФЯИАћЦї-КкЫиаЁЬх(melanosome),ЦфжагаКЌЭЕФЗгбѕЛЏУИ,МДРвАБЫсУИ(tyrosinase),ЫќПЩНЋРвАБЫсзЊБфГЩКкЩЋЫи(ЭМ4-23).АзЛЏВЁШЫгаКкЫиЯИАћ,ЕЋРвАБЫсУИШБЗІ,ЪЙКкЩЋЫиВЛФмаЮГЩ,ВЁШЫвђШБКкЩЋЫиЖјАзЛЏ.АзЛЏВЁДцдквХДЋвьжЪад.вбжЊАзЛЏВЁжСЩйга7жжВЛЭЌРраЭ.РвАБЫсУИЛљвђ(TYR)ЖЈЮЛгк11q14-q22.
ЁЁЁЁЪєгкДЫРрЕФР§згЩагавХДЋадМззДЯйжз,ЫќЪЧгЩгкХМСЊУИЁЂЭбЕтУИЛђЕтЙ§бѕЛЏУИШБЗІЕМжТМззДЯйЫиЩњГЩМѕЩйв§Ц№ЕФ,ЛМепЩэВФАЋаЁ,жЧСІЕЭЯТ,УцУВГѓТЊ.
ЁЁЁЁ(4)УИШБЗІжТХдТЗВњЮядіЖрв§Ц№ЕФМВВЁ:ЕБУИЕФШБЗІЕМжТжївЊДњаЛЭООЖЪмзшЖЯЪБ,Й§СПЕФЧАЬхЮяS2ЭЈЙ§СэвЛХдТЗДњаЛв§Ц№ФГаЉИБВњЮяЕФЖбЛ§.ШчЙћдіЖрЕФХдТЗВњЮяЪЧЮоЖОЕФ,дђПЩХХГіЬхЭтВЛжТЮЃКІШЫЬх,ЕЋШчЙћХдТЗВњЮяЛђЦфЗжНтВњЮягаЖО,дђПЩЮЃКІЛњЬхв§Ц№МВВЁ.
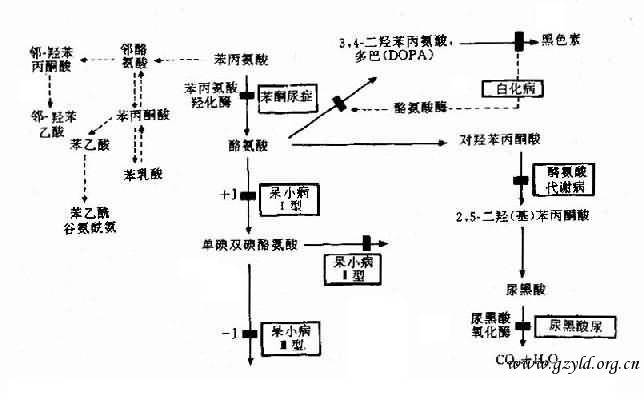
ЭМ4Ѓ23 БНЯжАБЫсДњаЛМАгаЙиЕФвХДЋадУИВЁЕФЗЂВЁЛњРэ
ЁЁЁЁБНЭЊФђжЂЃЈphenylketouria,PKUЃЉПЩзїЮЊЪЕР§ЁЃБОВЁвджЧФмЗЂг§ВЛШЋЮЊжївЊЬиеїЃЌГЪГЃШОЩЋЬхвўадвХДЋЃЌШКЬхЗЂВЁТЪдМ1ЃЏ16000ЃЌгЩБНБћАБЫсєЧЛЏУИЃЈphenylalanine hydroxylase,PAHЃЉвХДЋадШБЗІв§Ц№ЁЃЯжвбжЊБНБћАБЫсєЧЛЏУИЛљвђЖЈЮЛгк12q24.1,ДЫЛљвђШЋГЄдМ90kbЃЌКЌ13ИіЭтЯдзгЃЌдкжаЙњШЫжавбЗЂЯж10гржжЕуЭЛБфЃЌетЪЧдьГЩУИЛюадШБЗІЕФдвђЁЃ
ЁЁЁЁЕфаЭPKUЛМЖљГіЩњЪБЃЌЭтУВе§ГЃЃЌдМжС3Ѓ4ИідТЪБЃЌНЅГіЯжжЧФмЗЂг§ВЛШЋЃЌЛМЖљВНЗЅаЁЃЌзЫЫЦдГКяЃЌМЁеХСІдіИпЃЌвзМЄЖЏЃЌЩѕжСОЊиЪЃЌ90ЃЅвдЩЯЛМепУЋЗЂЗЂЛЦЃЌЗєАзЃЌЩѕжСКчФЄГЪЛЦЩЋЃЈАзжжШЫГЪРЖЩЋЃЉЁЃДЫЭтЃЌЛМЖљФђКЭКЙга ЬиЪтЕФИЏГєЁЃ
ЬиЪтЕФИЏГєЁЃ
ЁЁЁЁе§ГЃШЫБНБћАБЫсдкЬхФкжївЊЭЈЙ§PAHЕФзїгУзЊБфГЩЮЊРвАБЫсЃЌМЬЖјЩњГЩКкЩЋЫиЃЈЭМ4Ѓ23ЃЉЁЃPAHжївЊДцдкгкИЮжаЃЌЫќЕФзїгУашвЊИЈвђзгЫФЧтЩњЮяЕћрЄЃЈBH4ЃЉЃЌШчЙћPAHШБЗІЃЈЛюадЃМ1ЃЏ10ЃЉПЩзшЖЯБНБћАБЫсзЊЛЏЮЊРвАБЫсЃЌМДВњЩњЕфаЭЕФБНЭЊФђжЂЁЃУИЛюадВПЗжШБЗІЃЌПЩЕМжТЧсЖШИпБНБћАБЫсбЊжЂЃЈhyperphenylalaninemiaЃЉЁЃ